19. Synthesis of Furanones bearing alpha-Chiral Quaternary Stereogenic Center
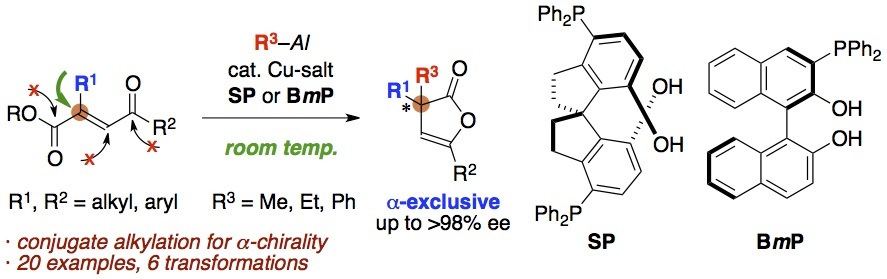
“Highly Chemo-, Enantio-, and Regioselective Synthesis of α,α-Disubstituted Furanones via Cu-Catalyzed Conjugate Addition”
Endo, K.*; Yakeishi, S.; Takayama, R.; Shibata, T.*
Chem. Eur. J. 2014, 20, 8893–8897.
1年半以上かかって,ようやく登場.金沢の学生が2ヶ月くらいで放棄したテーマという曰く付き.
ちなみに,まだ図がありませんので,そのうち更新します.
フラノンとは.要するにbutenolideの異性体で,要するにbutenolide.ただ単に,カルボニル部位を強調したいので,フラノンと呼んでみただけであったりする.何が違うかというと,butenolideの多くはカルボニルのγ位に置換基を持ち,共役エノン構造をとっている.一方のフラノンは,カルボニルα位に置換基があり,共役エノン構造ではない.
ただし,α位のモノアルキル化フラノンは自発的に異性化して,ここで言うところのbutenolideになる可能性が高い.だからカルボニルα位をジアルキル化などして4級炭素としてやらねば,安定に得ることが難しいと思われる.
そんなフラノンの立体選択的合成法は,これまでに2つしか報告されていない.脱炭酸アリル化と,アシル移動反応である.エナンチオ選択性がベストでも95% eeを越えないという中途半端加減は良いとしても,これらの反応は位置選択性が微妙である.つまり,α位のアリル(アシル)化だけではなく,異性化した挙げ句にγ位のアリル(アシル)化も競合する.その比率は,α:γ=1:1から10:1程度まで.それほど良い結果ではない.2つしか合成法がない上,位置選択性,エナンチオ選択性,いずれも中途半端,という歴史がある.
では,フラノンは何に使えるのか?…実は,明確な合成的利点は知られていない.まずbutenolideは,天然物にも含まれる骨格なので,その有用性は,かなり高い.butenolideの誘導化により全合成にも展開されているし,合成法も多い.フラノンは合成法が少ないし,その用途もあまり知られていないが,この化合物のポテンシャルは高い.なぜターゲットにならなかったか曖昧だ.α位に4級不斉を有するカルボニル化合物は,多様な場面で登場する,真に有用な合成ビルディングブロックである.そのカルボニル部位がエステルとなっている(正確にはlactoneか)フラノンは,さらに酸素原子に隣接する二重結合を有する.とにかく,色々な分子変換の鍵となる官能基に富んでいるのである.
しかし,合成が難しい.なぜか.それはフラノンからエノラートを発生させようという発想が既に障壁となっているからだ.フラノン由来のエノラートの求核部位は,α位とγ位.これを完全に制御しようというのは厳しい.つまり,エノラートを調製してから求電子剤と反応させようという常識的なアプローチでは駄目ということになる.
ということで,ラクトン化でフラノンを合成すれば良いことになる.あまり細かく書くとネタが拡散してしまうが,ここでは,共役付加により生じるエノラートの酸素原子を分子内でエステル部位に付加させてラクトン化して,フラノンに誘導すれば良いという発想になる.実に単純であった.
が,この単純な発想に,案外,気づかないらしい.我々が本研究に取り組んでいる最中に,Hoveyda教授らが彼らの独自触媒を用いる非環状エノンへの共役付加反応により,4級不斉構築するという報告をした.この論文で,彼らは,今回登場する基質であるケトエステルを用いている.はっきり言って,触媒が違うだけで反応そのものは同じなのだが,彼らは,生成物が,非環状のケトエステルとして得られると述べている.なお,収率は67%で選択性が82% eeなので,しっかり制御できたとは言えない.彼らのケースでは,–30 °Cという低温条件で反応させている.おそらく立体選択性が大きく低下するのがネックなのだと思われるが,その温度条件では,単に,生成物のエノラートからラクトン化が進行しないという結論で良いのかもしれない.本件について,2016年11月25日にHoveyda教授の講演前のディスカッションで質問してみたところ,彼らは,やはりフラノンは得られておらず,たぶん温度条件の違いが効いているのでは,と言っていた.他にも要点はある気はしているが,その話は,またいずれ.
我々のケースでは室温でも高いエナンチオ選択性が維持できるため,わざわざ低温にする必要がない.ということで,条件を色々と検討してみたところ,ベンジルエステルを用い,酢酸銅とSPINOL-PHOSを触媒前駆体とすれば,目的のフラノンのみが,位置選択的に,88%収率97% ee程度で得られることが明らかになった.まずはメチル化でスクリーニングしている.最高で99% eeで生成物を得ることができる.
メチル化といえば,合成的には利用価値が高い.長居アルキル基を導入しても,不斉反応では,そんなに意味はない.エチル基くらいなら天然物にも,少しは存在するのだが,長いアルキル鎖は冗長で無駄ともいえる.だけど,メチル化だけでは,評価は下がる…要するに,汎用性が低いと判断されるわけだ.
そこでエチル化した.素のままの条件では,反応時間が長く,エナンチオ選択性は80% eeを越えるくらいだった.これは遷移状態に関与するアルミニウム錯体のLewis酸性に依存していると考えられる.だから,Lewis酸を添加した.その結果,eeこそ,それほど向上していないが反応時間は1/10くらいになった.Lewis酸の効果が劇的であることはわかったが,主旨ではないので,今回は深追いしていない.
では,本反応の遷移状態モデルを見てみよう.R3AlとCuXのトランスメタル化で生じるR2AlXとCuRが鍵だ.R2AlXのAl原子がケトエステルのケトン部位を活性化し,そのXが銅原子に配位しつつ遷移状態へと誘導している.すなわち,R = Meの場合はLeiws酸性が十分であったが,R = EtになるとLewis酸性が低下してカルボニル基の活性化が不十分になってしまったといえる.だからこそのLewis酸添加剤の効果である.
一方で,フェニル化も検討してみた.我々の触媒システムでは,これまでフェニル化を本格的には試してこなかった.今回は,いずれにせよ汎用性を広げるためには避けて通れないため,ここで一発,手法として確立しようという意気込みになった.
さて問題は,試薬である.Me3AlとEt3Alくらいなら市販品だが,フェニルアルミニウムの誘導体は簡単に入手できない.そのため,調製しなければならないわけだ.難しいわけではないが,フェニル化の試薬として考えられるものがPh3Alか,PhR2Alである.前者は塩化アルミニウムとフェニルリチウムかフェニルグリニアから調製可能で,後者は R2AlClとフェニルリチウムかフェニルグリニアから調製可能.
まずは,Me3Alなどに倣い,Ph3Alを試してみたが,反応は進行しないという結果になってしまった.
一方で,PhMe2Alを用いると,目的となる生成物がCu(OTf)2を触媒前駆体としたとき,高エナンチオ選択的に得られることを見いだした.なお,Me化された生成物も17%程度で得られる.トルエンを展開溶媒としてカラム精製すると分離できる.
さて,フラノンの変換法について,いくつか試してみた.フリーデルクラフツ型の反応では,カルボニルに付加せず,オレフィン部位に付加した生成物のみが得られる.けっこうかさ高そうだが.
インジウム触媒を用いてアミド化すると,ラクタムが得られる.ラクトンからラクタムに変換するのは案外難しい.
水和するとγ-ケト酸が得られ,水素還元すると脱ベンジルを伴い非環状のトリアルキル型カルボン酸になる.基質次第では,ラクトンが得られるが,収率が低いか,ジアステレオ選択性が低い.
最後にアルデヒドに還元して分子内でアルドール縮合すると,天然物の合成中間体が得られる.
以上,フラノンの高立体選択的な合成法を初めて開発した.化学選択的に,3級不斉より4級不斉を好んで構築するという変わり種である.ちなみに,我々の複核金属錯体触媒は,既存の触媒と比較すると,高い反応性を示す基質が異なることは,なんとなくわかっている.そのため,おそらく,他の触媒では同様の結果になることは今のところないかなと感じている.唯一,Hoveyda教授らが競合しているが,彼らの触媒とも触媒活性の傾向は異なる.彼らは彼らの独自触媒なので,正確には競合という感覚はない.
まだいくつかのネタで,不斉共役付加反応の論文が出る予定.検討したいものは多くあっても人手は足りず.
そろそろ,新しい配位子を合成して,色々な反応に用いていきたいものである.